
Du Banc À la Clinique – Chronologie des Études Précliniques d’Innocuité
Introduction
Le processus de développement de médicaments comporte de nombreuses phases qui peuvent prendre de 12 à 15 ans et coûter plus d’un milliard de dollars. Il est essentiel d’avoir une feuille de route et un plan bien développés à l’esprit dès les premières étapes du développement afin de minimiser la quantité de ressources gaspillées tout au long du processus.
Commencer le processus avec la fin en tête est la clé d’un processus de développement fluide et efficace.
Dans ce bulletin, nous donnerons un aperçu général des tests courants effectués au cours du processus de développement de médicaments, du banc d’essai à la clinique. Nous discuterons brièvement du processus de découverte et de sélection des candidats principaux, puis nous décrirons certains des tests de sécurité précliniques courants requis pour soumettre la documentation requise pour l’approbation réglementaire des essais cliniques. Enfin, nous présenterons le rôle d’ITR dans ce processus.
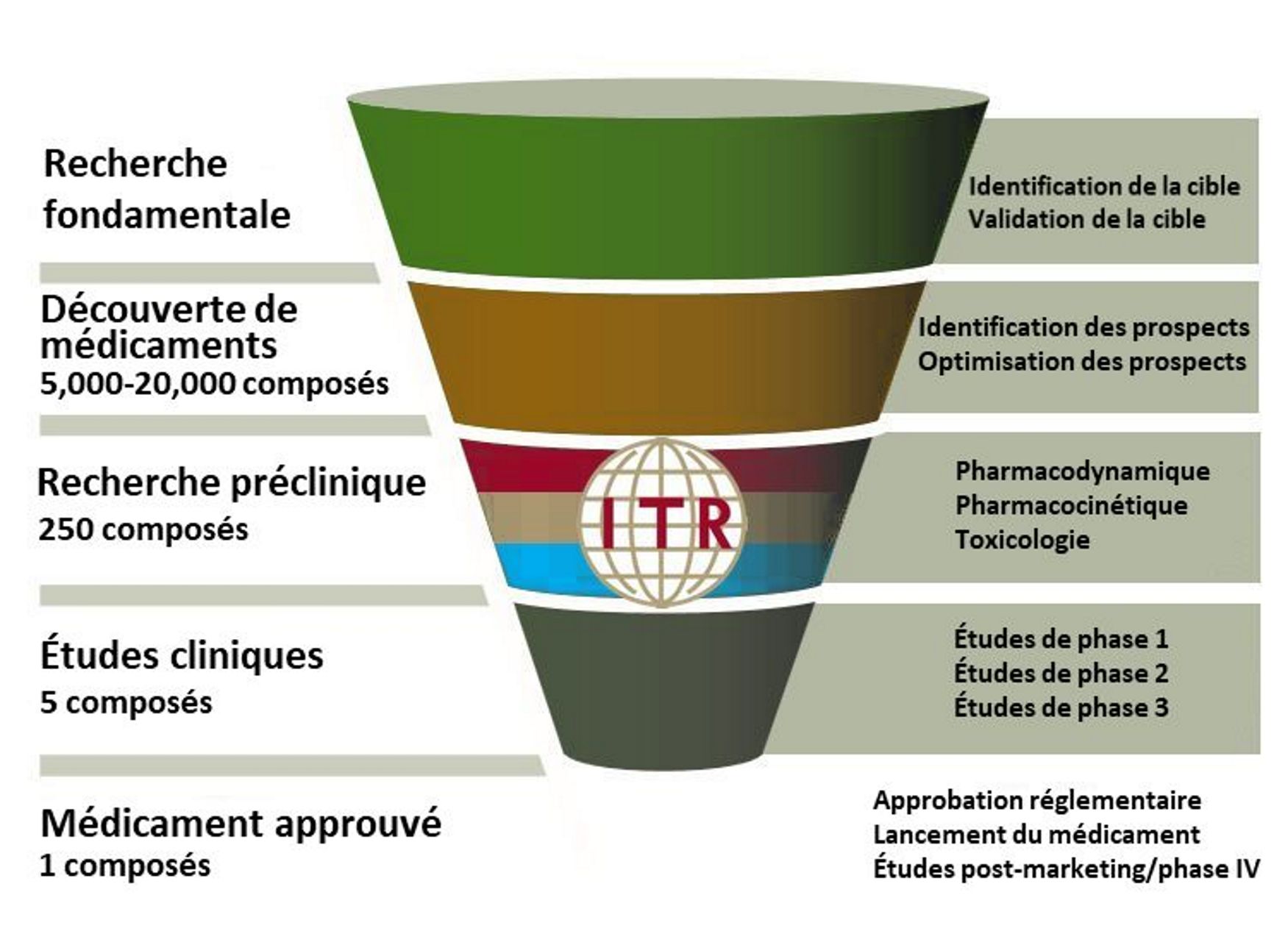
Avant ITR : Sélection du Candidat Principal et Efficacité Préclinique
Les premières phases du développement de médicaments comprennent la découverte de médicaments, la sélection de candidats principaux et l’efficacité préclinique.
Le processus de découverte commence par l’identification d’une cible médicamenteuse. Ce processus est assisté par l’utilisation de la bioinformatique, du criblage phénotypique et des études d’association génétique. Des tests sont effectués pour déterminer si altère la fonction de la cible avec un bénéfice thérapeutique pour le patient.
Le criblage à haut débit teste l’interaction entre les composés et des lignées cellulaires sélectionnées ou des protéines cibles. Un grand nombre de composés potentiels sont rapidement criblés jusqu’à ce que celui qui se lie avec une haute affinité à la cible biologique soit identifié comme nouveau médicament potentiel. Alternativement, un criblage à faible débit avec des composés présélectionnés est effectué sur des animaux, des tissus ou des organes. Les méthodes à faible débit sont des procédures plus détaillées, mais nécessitent plus de temps. Au moment où un candidat principal est identifié, des milliers de composés auront été testés et rejetés.
Le composé principal sera sélectionné pour un certain nombre de critères souhaités : spécificité, puissance, sélectivité. Le composé doit avoir un mode d’action défini, il doit avoir une forte affinité pour la cible souhaitée ainsi qu’une sélectivité pour la cible visée par rapport aux autres cibles potentielles. D’autres tests in vivo sont également effectués pour recueillir des données sur l’absorption, la distribution, le métabolisme et l’excrétion (ADME). De nombreux candidats-médicaments sont éliminés du développement ultérieur en raison de mauvais résultats pharmacocinétiques (par exemple, faible biodisponibilité, demi-vie très courte, etc.). En tant que tel, ce dépistage précoce réduit les risques de rejet plus tard dans le cycle de développement.
Une fois que le composé principal a été sélectionné, que la voie et la fréquence d’administration ont été identifiées et que l’efficacité a été démontrée in vivo et/ou in vitro, l’étape suivante consiste à s’assurer que le composé est sans danger pour l’administration chez l’homme. Ceci est accompli grâce à une série d’études de sécurité précliniques non conformes aux BPL et aux BPL.
Dose Maximale Tolérée, Pharmacocinétique et Détermination de la Plage de Doses
À la fin des études primaires de pharmacologie/d’efficacité, l’étape suivante consiste à déterminer l’innocuité du composé en commençant par la détermination de la dose maximale tolérée (MTD).
La DMT est la dose la plus élevée d’un médicament qui ne provoque pas d’effets secondaires inacceptables ou de toxicité manifeste.
Les études MTD sont généralement des études à dose unique qui utilisent un schéma posologique croissant chez un petit nombre d’animaux. La dose initiale est généralement égale ou légèrement supérieure à la dose thérapeutique/efficace prévue. Les doses suivantes sont augmentées progressivement au fil du temps jusqu’à ce que des signes cliniques légers à modérés soient observés.
Le choix des espèces animales dépend de la nature du candidat médicament. Pour les médicaments à petites molécules, les rats et les chiens sont les plus couramment utilisés. Pour les grosses molécules, une ou deux espèces dans lesquelles l’élément d’essai est pharmacologiquement actif doivent être sélectionnées en raison de l’expression du récepteur ou d’un épitope.
Les données pharmacocinétiques (PK) permettent aux chercheurs de comprendre le devenir du médicament dans le système biologique. Les études pharmacocinétiques consistent à collecter des échantillons de sang en série au fil du temps, puis à analyser les échantillons pour déterminer les niveaux de concentration de médicament dans le sang, le plasma ou le sérum. Des études pharmacocinétiques sont également réalisées sur un petit nombre d’animaux.
Les données recueillies à partir des études MTD et PK sont ensuite référencées pour permettre la sélection des niveaux de dose et du schéma posologique pour les études de détermination de la plage de doses. Les études de recherche de gamme utilisent une conception multi-groupes avec un groupe témoin, un groupe à faible dose, un groupe à dose moyenne et un groupe à dose élevée et durent généralement de 7 à 14 jours. Les résultats obtenus à partir de ces études permettent de sélectionner les niveaux de dose pour les études définitives de toxicité à doses répétées.

Autres Études Complémentaires : Pharmacologie de Sécurité, Tolérance et Toxicologie Génétique
Des tests de dépistage précoces et des tests pour déterminer les informations posologiques peuvent être effectués dans des conditions non conformes aux BPL, mais le passage d’un médicament de la phase préclinique à la clinique nécessite une liste d’études de sécurité BPL, y compris la pharmacologie de sécurité, la toxicologie génétique et des études de toxicité à doses répétées à plus long terme.
Les tests pharmacologiques d’innocuité sont destinés à dépister les effets indésirables sur les systèmes d’organes vitaux, tels que les systèmes cardiovasculaire, respiratoire, rénal, gastro-intestinal et nerveux central.
Les études de tolérance sont réalisées en utilisant la méthode d’administration prévue et ne sont pas toujours réalisées avec chaque médicament. Des études de tolérance locale sont souvent réalisées lorsque le médicament est destiné à être administré par voie topique sur la peau ou les yeux.
Alors que certains tests de dépistage de toxicologie génétique peuvent être effectués dans des conditions non conformes aux BPL plus près de la phase de découverte, les tests de toxicologie génétique BPL sont requis dans le cadre de la batterie de tests pour les soumissions IND.
Des tests de toxicité génétique sont effectués pour évaluer les dommages chromosomiques potentiels ou la mutagénicité.
Les tests de toxicité génétique consistent en des tests in vitro, tels que le test d’Ames, l’aberration chromosomique et le test du micronoyau in vitro, ainsi que des tests in vivo, tels que le test du micronoyau in vivo.

Études À Doses Répétées
Une fois que le nouveau candidat-médicament a subi des tests d’innocuité dans des études à dose unique, des études à doses répétées sont réalisées dans des conditions BPL. Ces études sont destinées à évaluer les effets toxiques potentiels selon le schéma posologique clinique et la méthode d’administration prévus et, par conséquent, les études à doses répétées doivent imiter autant que possible l’utilisation clinique prévue.
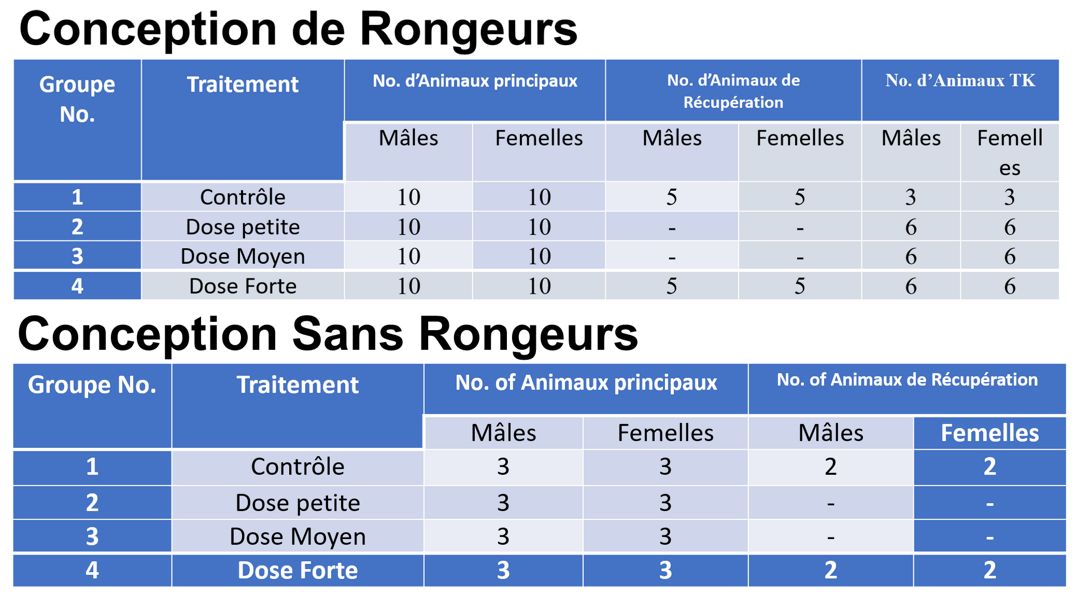
Les études à doses répétées sont réalisées en utilisant un nombre relativement plus important d’animaux et la conception de l’étude est définie par les résultats des études MTD, PK et de recherche de gamme. Ces études sont généralement réalisées sur une espèce de rongeurs ainsi que sur une espèce de non-rongeurs et une conception d’étude typique aura un dosage pendant 28 jours.
Vous trouverez ci-dessous des exemples de conceptions d’études chez les rongeurs et les non-rongeurs. Les études à doses répétées auront toujours un groupe témoin pour les comparaisons statistiques. Une cohorte d’animaux en rétablissement est également généralement incluse dans les études définitives de toxicité à doses répétées afin d’évaluer l’apparition retardée, la réversibilité ou la persistance des effets toxiques après l’arrêt de l’administration.

Ce Que Propose ITR
ITR est une organisation de recherche sous contrat en toxicologie préclinique qui soutient l’industrie biopharmaceutique en fournissant une large gamme de services non cliniques allant des études pharmacocinétiques à dose unique aux essais biologiques de cancérogénicité sur 2 ans chez diverses espèces, notamment les souris, les rats, les cochons nains, les lapins et les chiens. et les singes. En plus de la batterie de tests standard requise pour les tests de sécurité des petites molécules, nous proposons également une large gamme de services pour le développement de grandes molécules dans nos espèces disponibles.
Depuis 1993, ITR est accrédité par le CCPA (Conseil canadien de protection des animaux) et l’AAALAC (Association pour l’évaluation et l’accréditation des soins aux animaux de laboratoire). Tous nos travaux sont guidés par des procédures opérationnelles standard (SOP) et entièrement conformes aux BPL (bonnes pratiques de laboratoire), y compris nos études non BPL.
ITR propose également des services pour tous les modes d’administration. Établie en tant qu’ORC axée sur la perfusion, ITR s’est diversifiée au cours des 30 dernières années, ajoutant une expertise de pointe en inhalation à notre portefeuille en plus de la toxicologie générale, des tests cutanés et oculaires.

Conclusion
Le développement de médicaments est un long parcours depuis l’identification initiale de la cible et la sélection du candidat principal jusqu’aux tests préliminaires d’efficacité et d’innocuité. Chez ITR, nous continuons à développer notre expertise en matière de tests de sécurité pour répondre à un plus large éventail de nouveaux candidats-médicaments et pour nous assurer que les sponsors reçoivent des informations de sécurité plus complètes sur leurs candidats-médicaments. Qu’il s’agisse de petites molécules ou de grosses molécules, ITR aide nos sponsors à faire passer rapidement leurs candidats-médicaments du banc d’essai à la clinique.
